If You Eat Excess Protein, Does It Turn Into Excess Glucose?
Gluconeogenesis is Demand-Driven, not Supply-Driven
We have seen the claim that any protein you eat in excess of your immediate needs will be turned into glucose by spontaneous gluconeogenesis 1. (Gluconeogenesis (GNG) is the process by which glucose is made out of protein in the liver and kidneys.) Some people think that because protein can be turned into glucose, it will, once other needs are taken care of, and that therefore keto dieters should be careful not to eat too much protein.
While we believe there are valid reasons for limiting protein intake, experimental evidence does not support this one. In our opinion, it makes sense physiologically for GNG to be a demand-driven rather than supply-driven process, because of the need to keep blood glucose within tight bounds.
In brief
- Gluconeogenesis is a slow process and the rate doesn’t change much even under a wide range of conditions.
- The hypothesis that the rate of gluconeogenesis is primarily regulated by the amount of available material, e.g. amino acids, has not been supported by experiment. Having insufficient material available for gluconeogenesis will obviously limit the rate, but in the experiments we reviewed, having excess material did not increase the rate.
- We haven’t found any solid evidence to support the idea that excess protein is turned into glucose.
- More experiments are needed to confirm that this still holds true in keto dieters.
Gluconeogenesis has a Stable Rate
Gluconeogenesis (GNG) is a carefully regulated process for increasing blood sugar.
It is stimulated by different hormones, including glucagon — the primary hormone responsible for preventing low blood sugar. GNG produces glucose slowly and evenly 2. It was once thought that the main determination of the rate of GNG was how much glucogenic substrate, that is, raw materials for it, was available, but further experiments have shown that this is not the case 3. Instead, it now appears that GNG is relatively constant over a large variety of conditions 4.
As an example of this stability, a study by Bisschop et al. in 2000 5 showed that subjects following a keto diet for 11 days had only a small (14%) increase in glucose production from GNG after overnight fasting, as shown in this graph. This works out to a difference of less than a gram of glucose per hour.
Note that 11 days might be too little time for all of the subjects to keto-adapt, and it is possible that the rate of GNG would change in subsequent weeks.
Negative Results
In another experiment (this time in subjects on a glycolytic, or carb-based, rather than a ketogenic diet), ingesting 50g of protein resulted in the same amount of glucose production as drinking water 6. In other words, the amount of glucose that was made after ingesting that protein wasn’t any more than would have been produced without it. While it’s possible that this protein doesn’t count as “excess”, it was likely to be nearly half of their daily required protein intake, and eaten in one sitting, and so it is enough to cast serious doubt on the idea.
There are other experiments in which increasing the available material for GNG to high levels didn’t increase GNG 3, 4. In these experiments GNG substrates were infused directly into the blood rather than eaten.
The problem with applying the results of these experiments to the question of excess protein consumption is that infusion might bypass some mechanism that increases GNG when the protein is actually eaten. For instance, it is known that protein consumption stimulates a great deal of glucagon (along with insulin) 7, and it might be suggested that this glucagon would thereby increase GNG. A counterargument to that possibility is that although glucagon stimulates GNG in many conditions, its action appears to always be overridden by insulin 8. This means that the insulin that is produced when eating protein will counteract the glucagon and GNG will not be affected (except in the case of insulin-dependent diabetes, where insulin is neither created nor responded to in the normal fashion).
Both the argument from infused substrates and the counter-arguments outlined here are plausible mechanism arguments — taking physiological processes known to occur in one context and arguing that they will occur in another context. Plausible mechanism arguments should be used with caution.
Summary
In sum, then, there is no evidence that we could find that consuming excess protein will increase glucose production from GNG. On the other hand, there is much suggestive evidence that it does not.
Further experiments need to be carried out to answer the question completely. In particular, we would like to see a comparison of the rate of GNG in keto-adapted dieters consuming no protein, adequate protein, or a large quantity of protein, with and without dietary fat.
Follow-up posts
For clarification and further discussion of this topic, please see:
- We were talking about gluconeogenesis, not ketogenesis.
- Protein, Gluconeogenesis, and Blood Sugar
- Protein, Ketogenesis, and Glucose Oxidation
References
(We owe a debt of gratitude to a special friend from Windy City for helping us access full texts, as our previous access has expired. Thank you!)
1. Evidence type: observation
Please note:
We have cited some people here as making what we believe to be an unsupported assertion. This does not imply any disrespect for the authors! To the contrary, we believe that writers such as these contribute to scientific knowledge even when they make mistakes. By writing specific and falsifiable statements and by posting them publicly where others can cite them, they give others a chance to learn from both their accurate statements and their mistakes.
We, too are fallible, and we expect that errors of our own will come to light sooner or later; such is the nature of science. There is no shame in this, and we intend none. Please see Apologia for our philosophy about this.
Scientific progress is made in large part through discovering errors and correcting them. We sincerely hope that those we have quoted will be glad to either learn from this post, or conversely, to point out to us where we have erred. In either case, an issue that was obscure will have been clarified for everyone.
Nora Gedgaudas
Also, keep in mind that a significant percentage of protein consumed that is in excess of what you actually need for your daily maintenance and repair will convert to sugar and get used exactly the same way.
As I’ve said before, I’m trying to minimize my use of glucose, whether exogenous or endogenously produced. If I’m eating so much protein that the excess is being converted to glucose, I’m not really minimizing it, am I?
Eric Westman in an interview with Jimmy Moore
34:37
JM: Well, and I would think that if you’re sensitive to carbohydrate then you would be sensitive to eating too much protein as well, because you want to stave off the effects of gluconeogenesis from happening, which would provide too much glucose in your body, tantamount to eating a lot of carbs.
EW: That’s a good point, that some of the protein that we eat can be turned into the glucose through gluconeogenesis, and that may be a reason why someone is not able to get to ketosis — that too much protein is being converted to glucose.
(Update 2012-08-21)
When you eat more protein than your body needs to replace and repair body parts, excess protein is largely converted into glucose and burned as fuel.
2. Evidence type: experimental
(Emphasis ours)
In the process of protein metabolism, the complex protein molecule is split in the intestinal tract to amino-acids. These are absorbed into the blood stream and transported to the liver where oxidative deamination occurs. Here the glycogenic amino-acids are split to form urea and glucose. That this process is a slow one is shown in the charts by the slowly rising blood urea nitrogen. Glucose is, therefore, liberated into the blood stream in this process at a slow and even rate over a prolonged period of time. Under these conditions the diabetic is able to utilize a greater total amount of glucose without glycosuria in the eight hour period. Therefore, the inability of a diabetic to dispose of large quantities of glucose is partially compensated if the glucose is presented for utilization slowly and evenly. There appears, then, to be some advantage to the diabetic of this slow liberation of glucose from protein foods.
3. Evidence type: review of experiments
(Emphasis ours)
Gluconeogenesis plays an integral role in the maintenance of glucose homeostasis in humans, contributing about one-third of glucose produced in the postabsorptive state and all glucose produced when hepatic glycogen is depleted by starvation (6, 23-25). Because the results of in vivo experiments in humans and animals (12-15, 20) and in vitro perfused rat liver studies (11, 27) have demonstrated a close correlation between the rate of glucose production and the flux of gluconeogenic substrates, it is believed that gluconeogenic precursor supply plays a major role in the regulation of glucose production (12,13,20). Several studies in vivo support this concept. For example, we and others have demonstrated that the hyperglycemic response to severe burn injury and sepsis is a direct result of an increased rate of glucose production, which is associated with a concomitant increase in the fluxes of alanine and lactate, major gluconeogenic substrates (15, 39). The proposed regulatory role of precursor supply received further support in the quest to rationally explain the paradox of a reduced glucose production rate (and hypoglycemia) in starvation, despite a hormonal-substrate milieu that would normally favor stimulation of gluconeogenesis (2, 7, 12, 13, 28), thus glucose production. After prolonged starvation (3-4 wk), human subjects had low levels of gluconeogenic precursors associated with hypoglycemia and a reduced glucose production rate (6, 7, 12, 25). Infusion of unlabeled alanine caused hyperglycemia and an increased incorporation of [ 14C]label from alanine into glucose in this circumstance (12,13). It was therefore proposed by Cahill, Felig, and Marliss and their associates (7, 12, 13, 20) that the reduced glucose production rate in starvation was due to the reduced availability of gluconeogenic substrates; hence, gluconeogenic precursor supply was rate-limiting for glucose production rate.
In contrast, the findings of several kinetic studies performed in human and dog do not support this proposal (1, 30, 34, 38). These studies in postabsorptive subjects employed either the isotope dilution or hepatic vein catheterization techniques and failed to show any significant change in glucose production rate in response to infusions of substantial quantities of alanine, lactate, and glycerol even when there was a fivefold increase in the hepatic uptake of the infused substrate (1, 30, 34, 38)
These conflicting findings suggest that the relationship between gluconeogenic substrate supply and gluconeogenic enzyme activity in prolonged starvation may be different from that of the postabsorptive state. Alternatively, it is possible that the response to an increase in precursor supply is different from the response to a decrease. This latter possibility could occur if the endogenous supply of gluconeogenic precursors is just sufficient to maximally satisfy the capacity of the gluconeogenis enzyme system or of a particular key-limiting enzyme.
[…]
Our data so far indicate that under almost any physiological situation, an increase in gluconeogenic precursor supply alone will not drive glucose production to a higher level, suggesting that factors directly regulating the activity of the rate-limiting enzyme(s) of glucose production normally are the sole determinants of the rate of production; hence, there will be no increase in glucose production if the increase in gluconeogenic precursor supply occurred in the absence of stimulation of the gluconeogenic system. On the other hand, results of the DCA experiments suggest a coupling between precursor supply and gluconeogenic enzyme capacity. In this light, if there is a stimulation in gluconeogenic enzyme capacity (for example because of hyperglucagonemia of severe trauma), then there will have to be an increased rate of uptake of gluconeogenic precursors to meet the requirements of such a stimulated system. Thus the rate of uptake of gluconeogenic substrates and the rate of glucose production will be closely related, but the increased uptake of gluconeogenic precursors will be a consequence of a stimulated gluconeogenic enzyme system rather than the cause of an increased rate of gluconeogenesis.
4. Evidence type: review of experiments
(Emphasis ours)
Current data support the hypothesis that the rate of glucose appearance changes but the rate of gluconeogenesis remains remarkably stable in widely varying metabolic conditions in people without diabetes. In people with diabetes, whether gluconeogenesis remains unchanged is at present uncertain. Available data are very limited. The mechanism by which gluconeogenesis remains relatively constant, even in the setting of excess substrates, is not known. One interesting speculation is that gluconeogenic substrates substitute for each other depending on availability. Thus, the overall rate is either unaffected or only modestly changed. This requires further confirmation.
5. Evidence type: experimental
(Emphasis ours)
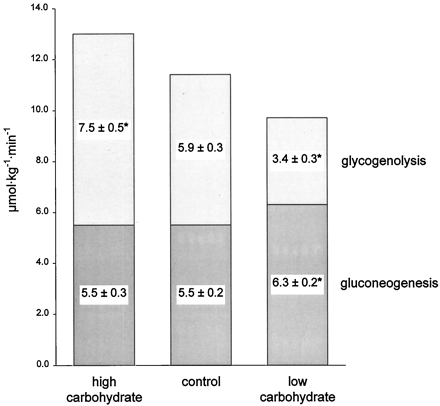
Abstract
To evaluate the effect of dietary carbohydrate content on postabsorptive glucose metabolism, we quantified gluconeogenesis and glycogenolysis after 11 days of high carbohydrate (85% carbohydrate), control (44% carbohydrate), and very low carbohydrate (2% carbohydrate) diets in six healthy men. Diets were eucaloric and provided 15% of energy as protein. Postabsorptive glucose production was measured by infusion of [6,6-2H2]glucose, and fractional gluconeogenesis was measured by ingestion of 2H2O. Postabsorptive glucose production rates were 13.0 ± 0.7, 11.4 ± 0.4, and 9.7 ± 0.4μ mol/kg·min after high carbohydrate, control, and very low carbohydrate diets, respectively (P < 0.001 among the three diets). Gluconeogenesis was about 14% higher after the very low carbohydrate diet (6.3 ± 0.2 μmol/kg·min; P = 0.001) compared to the control diet, but was not different between the high carbohydrate and control diets (5.5± 0.3 vs. 5.5 ± 0.2 μmol/kg·min). The rates of glycogenolysis were 7.5 ± 0.5, 5.9 ± 0.3, and 3.4± 0.3 μmol/kg·min, respectively (P < 0.001 among the three diets).
6 Evidence type: experimental
M A Khan, M C Gannon and F Q Nuttall. Glucose appearance rate following protein ingestion in normal subjects. J Am Coll Nutr December 1992 vol. 11 no. 6 701-706
Unfortunately, we have been unable to access the full text of this paper.
However, the results are described by the authors in the paper above (4) in text and in the table in the line marked [108]:
[T]here was no change in glucose production after ingestion of 50 g of protein in the form of cottage cheese.

If anyone having access to this paper would like to share it with us, we would be grateful, because it is the most relevant experiment we could find on the topic, and further details may be important.
7. Evidence type: experimental
(Emphasis ours)
Fasting glucose levels were 4.6 ± 0.2 mmol/l, and glucose levels did not change significantly during any of the tests. Fasting insulin levels were 55 ± 3 pmol/l. Insulin levels were unaltered after water ingestion, whereas they increased after fat and protein ingestion. The increased plasma insulin concentrations were seen between 30 and 240 min after fat ingestion (P = 0.031 vs. water) and between 15 and 240 min after protein ingestion (P = 0.018 vs. water). When compared with water ingestion, fat and protein ingestion both significantly increased early and late insulin responses (Table 1). These responses were more pronounced after protein than after fat ingestion (P < 0.001 for all). Fasting glucagon levels were 65 ± 3.7 ng/l. Glucagon levels were unaltered after water ingestion. In contrast, glucagon levels were increased by both fat and protein ingestion, with significant elevations from minute 120 and onward after fat ingestion (P = 0.019 vs. water) and from minute 30 and onward after protein ingestion (P = 0.005 vs. water). The late glucagon response was increased by fat ingestion, whereas, after protein ingestion, both early and late responses were significantly increased. As for insulin, early and late glucagon responses were higher after protein ingestion than after fat ingestion (both P < 0.001; Fig. 1).
8. Evidence type: review of experiments
(Emphasis ours)
Tracer studies in dogs have defined hormonal regulation of HGP [Hepatic Glucose Production] in detail. As in the isolated rodent liver, HGP is exquisitely sensitive to glucagon and insulin. Glucagon sets the basal tone, but insulin trumps glucagon at any concentration–just as it does in vitro. Both hormones affect primarily glycogenolysis by reciprocal changes of glycogen synthase and glycogen phosphorylase, and by modulating glycolysis through glucokinase, fructose-bisphosphatase and pyruvate kinase (see below) (Cherrington, 1999). Hormonal regulation of gluconeogenesis has proven difficult to demonstrate.